
Nucleic acid base pairs and ribosomal decoding fidelity and efficiency
IBMC > ARN > Eric Westhof
The twelve families of nucleic acid base pairs
The natural bases of nucleic acids form a great variety of base pairs with at least two hydrogen bonds between them. They are classified in twelve main families, with the Watson-Crick family being one of them. In a given family, some of the base pairs are isosteric between them, meaning that the positions and the distances between the C1’ carbon atoms are very similar. The isostericity of Watson-Crick pairs between the complementary bases forms the basis of RNA helices and of the resulting RNA secondary structure (covariation or coevolution). In addition, several defined suites of non-Watson-Crick base pairs assemble into RNA modules that form recurrent, rather regular, building blocks of the tertiary architecture of folded RNAs. RNA modules are intrinsic to RNA architecture are therefore disconnected from a biological function specifically attached to a RNA sequence. RNA modules occur in all kingdoms of life and in structured RNAs with diverse functions. Because of chemical and geometrical constraints, isostericity between non-Watson-Crick pairs is restricted and this leads to higher sequence conservation (neutral networks) in RNA modules with, consequently, greater difficulties in extracting 3D information from sequence analysis.
Tautomerism and recognition of nucleic acid base pairs
Nucleic acid helices are recognized in several biological processes like replication or translational decoding. In polymerases and the ribosomal decoding site, the recognition occurs on the minor groove sides of the helical fragments. With the use of alternative conformations, protonated or tautomeric forms of the bases, some base pairs with Watson-Crick-like geometries can form and be stabilized. Several of these pairs with Watson-Crick-like geometries extend the concept of isostericity beyond the number of isosteric pairs formed between complementary bases. These observations set therefore limits and constraints to geometric selection in molecular recognition of complementary Watson-Crick pairs for fidelity in replication and translation processes.
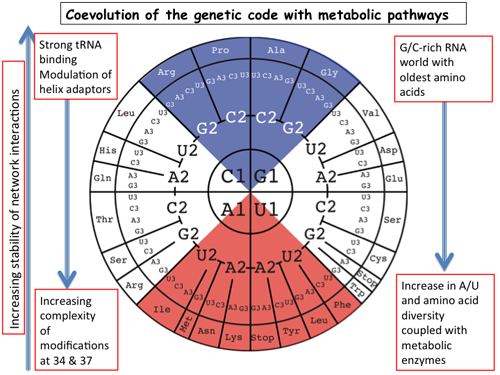
The ribosomal decoding site
The basic principles of mRNA decoding are conserved among all extant life forms. We present an integrative view all the complex interaction networks between mRNA, tRNA, and rRNA : the intrinsic stability of codon-anticodon trimers, the spatial conformation of the anticodon stem-loop of tRNA, the presence of modified nucleotides, the occurrence of non-Watson-Crick pairs in the codon-anticodon helix and the interactions with bases of rRNA at the decoding site. We derive an information-rich, alternative representation of the codon table. The new organization of the 64 codons is circular with an asymmetric distribution of codons that leads to a clear segregation between GC-rich 4-codon boxes and AU-rich 2:2-codon and 3:1-codon boxes. The advantage of integrating data in this circular decoding system is that all tRNA sequence variations can be visualized, within an internal structural and energy framework, for each organism and anticodon. Within this new representation, the multiplicity and complexity of nucleotide modifications, especially at positions 34 and 37 of the anticodon loop, segregate meaningfully and correlate well with the necessity to stabilize AU-rich codon-anticodon pairs and to avoid miscoding in split codon boxes. This structure-based network of interactions results in an energetically uniform decoding of all tRNAs that can adapt to the cellular constraints. The evolution and expansion of the genetic code is viewed as originally based on GC content with the progressive introduction of A/U together with tRNA modifications and the modification enzymes. This allows for a great diversity of codon usage depending on GC content of the genome and on the number and types of tRNAs. The representation should help the engineering of the genetic code to non-natural amino acids.
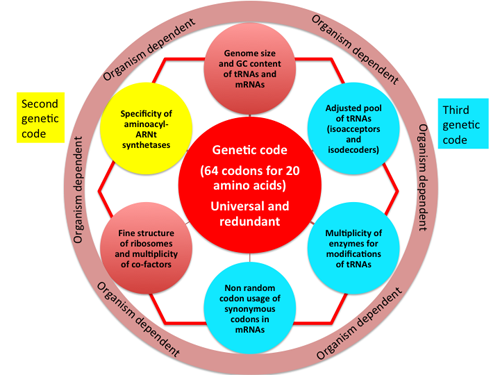
The cellular homeostasis and the central place of translation
In summary, in order to maximize the diversity in codon usage without increasing the number of different tRNAs (for decoding the 61 sense codons), cells developed sophisticated arrays of tRNA modifications that are anchored in cellular metabolic enzymatic pathways. The genetic code is not translated universally and several differences exist between organisms and in the three kingdoms of life. In each organism, there is a very strong connectivity between the elements responsible for the reliability and efficiency of the decoding process of the genetic code. The multiplicity of these highly interconnected elements and the integration of the various biological information flows ultimately allow for the maintenance of subtle cellular homeostasis and place the processes of translation at the center of cellular activities.
See in section Presentations (Download area and links) « RNA_Pairs_Tautomers_Modules.pdf »
See in section Download area and links the « Wheels for the genetic code for various organisms and cases ».
Selected publication :
- Grosjean, H., Westhof, E. (2016). An integrated, structure- and energy-based view of the genetic code. Nucleic Acids Research, 44, 8020-8040
- Westhof, E. (2014). Isostericity and tautomerism of base pairs in nucleic acids. FEBS Letters, 588, 2464-2469
- Rozov, A., Demeshkina, N., Westhof, E., Yusupov, M. & Yusupova, G. (2015). Structural insights into the translational infidelity mechanism. Nature Communications, 6, 7251
Professor Emeritus
